Protein misfolding that propagates and the mechanisms of neurodegenerative disease
Think of an organism—us, for instance—as a living construction project.
We all start with a blueprint: the unique combination of DNA molecules we inherit from our parents, a random combination of the DNA molecules that launched them a generation before.
With this blueprint laid out, the various types of RNAs take over. These are the engineering crews that take portions of the DNA blueprint and use the instructions to make proteins.
Proteins are the workhorses: the molecular machines, scaffolding, building blocks, and cellular messengers. The antibodies that help us fight disease, the hemoglobin that manages our blood’s acidity, the enzymes that facilitate a million different cellular reactions—these are all proteins, made with the instructions laid down in DNA and interpreted by RNAs.
Generating these many types of proteins is complicated. It starts by assembling long chains of amino acids. Molecules made up of carbon, oxygen, nitrogen, and hydrogen. The three-dimensional architecture of these complex molecules means that each contains some slightly magnetic “elbows,” portions of the structure with mildly positive or negative charges.
After the strings of amino acids are complete, some of these tiny charged spots on the various amino acids find each other and snap each chain into a wide variety of very specific folded shapes. Thanks to evolution, biological systems have found ways to use these variously folded proteins in unique and wonderful ways. They can form into tubes that let specific nutrients move safely into cells or create distinctive “keyholes” that can only be entered with a specific cellular key. They can even create proteins with a hydrophilic (water-loving) side while the other end prefers oil-based environments, and voila—a protein that acts like a fishing pole.
Figuring out how our many proteins fold is complex, and thousands of citizen scientists are now working on these problems, in a productive version of, say, Rubik’s Cube. We are fortunate to benefit from their puzzle addiction, as well as some new inroads in artificial intelligence, because it is pretty clear that misfolded proteins are involved in a lot of disease mechanisms, and researchers can use the help.
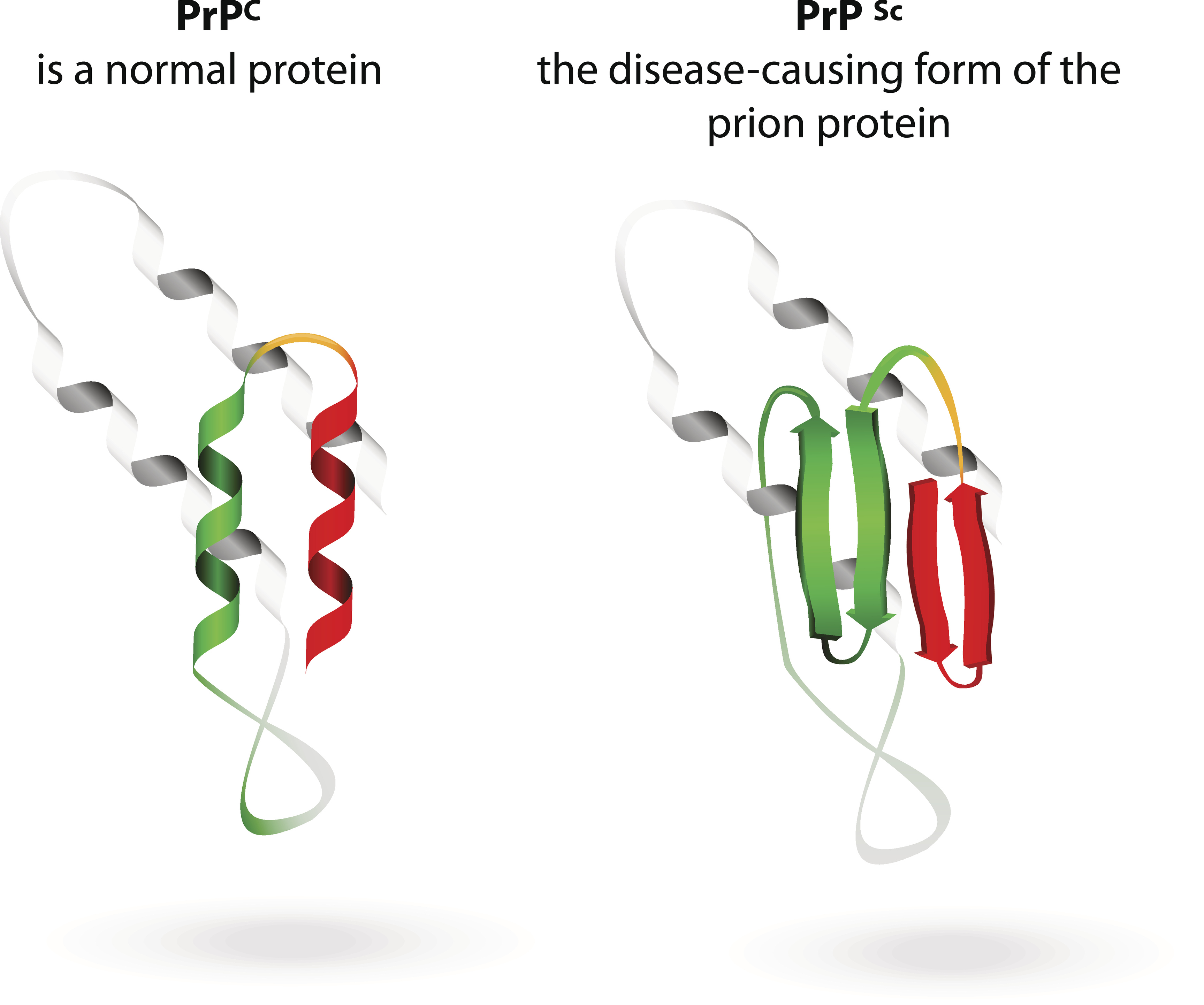
Among these are diseases caused by prions, misfolded proteins that can transmit their dysfunctional shape to normal proteins. Remember how much fun you had playing with your first Slinky? If it was metal (not the revised, plastic model), there always came a day when you would get that first kink or bend in your Slinky. Then it was just a matter of time before other parts of the coil started to kink up, and soon the whole thing was wrecked: the toy version of prion disease.
Several decades ago, there was an international furor about prion transmission. Some British cattle were found infected with “mad cow,” a deadly neurological disease, perhaps caused by feed made from sheep infected with scrapie, an ovine version of prion disease. (Both diseases impair walking and coordination and eventually lead to convulsive collapse and death in their animal hosts.) Other countries began refusing British beef, suspecting that eating the misfolded proteins in the cattle would cause the similarly horrible and deadly Creutzfeldt–Jakob disease, which attacks human brains. Since prion diseases can develop over a long period of time but kill quickly once established, beef became a somewhat scary menu item.
James Mastrianni, MD, PhD, studies these and other prion diseases—critically important work, as there is some evidence that many of the human neurodegenerative diseases, including Alzheimer’s, Parkinson’s, and ALS (Lou Gehrig’s disease), may all be caused at least in part by different kinds of misfolded proteins.
One method of attack in Mastrianni’s lab is to inject genetically vulnerable mice with different conformations of prions—a dangerous proposition that involves strict biosafety protocols to protect team members from accidental exposure. Mastrianni has developed an entire scoring system to evaluate and categorize motor behaviors in the mice as the prions cause analogous proteins to misfold.
His goal is to correlate the types of prion misfolding translate with specific motor problems, assess what happens when you delete sections of the misfolded protein and determine ways to induce normal folding. For instance, can one block interactions with the bad proteins? Or just block the particular segment within the protein that sets off the misfolding? What happens if you take out the errant genes and replace them?
In his busy neurological practice, Mastrianni applies what he learns from his studies of prion diseases in mice to patients with Alzheimer’s, dementia, and other neurological disorders, especially those who, like the mice, have identifiable genetic risks. Recently, he published a study of a woman in her 40s with progressive dementia; she also had a wobbly gait that started several years before. A deeper dive disclosed that other family members also suffered from early-onset dementia, and Mastrianni and his team found the woman had a mutation, rarely reported in African-American families like hers, which made her and her family especially vulnerable to prion disease. The woman died just over a year later, another tragedy in a family besieged with them.
Because the abnormal gene defect identified in this woman is so rare, Mastrianni is engineering a mouse with the same defect in hopes of better understanding how it makes carriers vulnerable to the disease. His lab is also developing “designer” gene therapies that target specific gene defects to shut them down and limit production of the misfolded protein.
Mastrianni hopes his work will change the prognosis for families like this woman’s, as well as diseases like Alzheimer’s—critical in our increasingly aging population.